
发布时间:2025-06-24 17:04:51 作者:北方职教升学中心 阅读量:603
注:主要讲述scanpy处理数据的结构、
通过conda,使用命令cd进入whl文件所在的目录后,然后通过pip安装:
pip installscanpy2. AnnData
2.1 AnnData的结构
在scanpy中,我们最常见的数据结构是AnnData,它是一个用于存储数据的对象,其数据结构可以描述如下: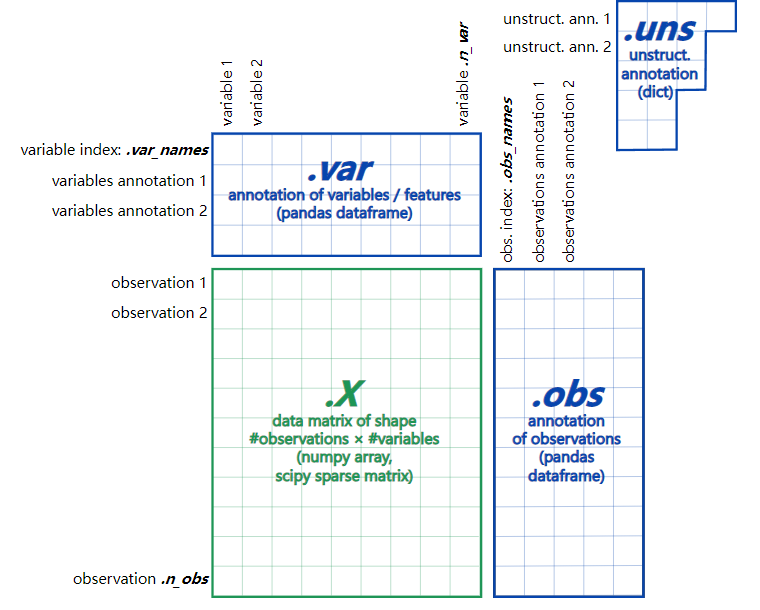
我们把上面这个对象记作 adata,我们需要了解以下几个部分:
| 功能 | 类型 | |
|---|---|---|
| adata.X | 矩阵 | numpy矩阵 |
| adata.obs | 观测量 | pandas Dataframe |
| adata.var | 特征量 | pandas Dataframe |
| adata.uns | 非结构化数据 | 字典dict |
为了进一步了解这个数据结构,我们手动构建一个AnnData对象:
importnumpy asnpimportpandas aspdimportanndata asadfromstring importascii_uppercase# 设置观测样本的数量n_obs=1000# obs用于保存观测量的信息obs=pd.DataFrame()# numpy.random.choice(a, size=None, p=None)# 从a(ndarray, 但必须是一维的)中随机抽取元素, 并组成指定大小(size)的数组# 数组p: 与数组a对应, 表示取数组a中每个元素的概率, 默认情况下选取每个元素的概率相同obs['time']=np.random.choice(['day1','day2','day4','day8'],size=n_obs)# 设置特征名var_namesprint(ascii_uppercase)# ABCDEFGHIJKLMNOPQRSTUVWXYZvar_names=[i*letter fori inrange(1,10)forletter inascii_uppercase]print(var_names)# ['A', 'B', 'C', 'D', 'E', 'F', 'G', 'H', 'I', ......, 'X', 'Y', 'Z',# ......# 'AAAAAAAAA', 'BBBBBBBBB', 'CCCCCCCCC', ......, 'YYYYYYYYY', 'ZZZZZZZZZ']# 特征数量n_vars=len(var_names)# 234# 将特征定义到 Dataframevar=pd.DataFrame(index=var_names)print(var.head())# 现在var没有columns(列索引), 只有index(行索引)# 创建数据矩阵 adata.XX=np.arange(n_obs*n_vars).reshape(n_obs,n_vars)1234567891011121314151617181920212223242526272829303132然后初始化 AnnData 对象,AnnData 对象默认采用数据类型 float32,我们为了便于后期观察打印结果,设置数据类型为 int32:
adata=ad.AnnData(X,obs=obs,var=var,dtype='int32')# 查看数据print(adata)"""AnnData object with n_obs × n_vars = 1000 × 234 obs: 'time'"""# 查看adata的X矩阵print(adata.X)"""[[ 0 1 2 ... 231 232 233] [ 234 235 236 ... 465 466 467] [ 468 469 470 ... 699 700 701] ... [233298 233299 233300 ... 233529 233530 233531] [233532 233533 233534 ... 233763 233764 233765] [233766 233767 233768 ... 233997 233998 233999]]"""1234567891011121314151617181920一般对于adata.X,行对应观测(即,细胞),列对应特征(即,基因);
我们每次操作 AnnData 时,并不是再新建一个 AnnData 来存储数据,而是直接找到已经在之前初始化好的 AnnData 的内存地址,通过内存地址来直接改变 AnnData 的值。
标记基因 (marker gene),是一种已知功能或已知序列的基因,能够起着特异性标记的作用。数据过滤(生信领域)和数据预处理(和机器学习类似,但是又有不同。这样做的好处是:
- 无需分配多余的内存;
- 可以直接修改已经初始化后的 AnnoData 对象;
比如:
# 查看 'A' 列的头三个元素print(adata[:3,'A'].X)"""[[ 0] [234] [468]]"""# 设置 'A' 列的头三个元素adata[:3,'A'].X =[0,0,0]# 再查看 'A' 列的头五个元素发现值被修改了print(adata[:5,'A'].X)"""[[ 0] [ 0] [ 0] [702] [936]]"""1234567891011121314151617181920但是,如果将 AnnData 对象中的一部分赋值到新对象,该对象会得到一块新内存用于存储实际数据,而不再是对原来adata对象的内存地址引用,比如:
adata_subset =adata[:5,['A','B']]print(adata_subset)"""View of AnnData object with n_obs × n_vars = 5 × 2 obs: 'time'"""# 为新对象 adata_subset 增加观测量 'foo'adata_subset.obs['foo']=range(5)print(adata_subset)"""AnnData object with n_obs × n_vars = 5 × 2 obs: 'time', 'foo'"""12345678910111213142.2 h5ad:AnnData的写入和读取
我们可以将AnnData对象通过h5ad文件保存到磁盘中,保存过程如下:
# 计算对象的大小defprint_size_in_MB(x):print('{:.3} MB'.format(x.__sizeof__()/1e6))# 查看对象大小print_size_in_MB(adata)# 0.187 MB# 查看是否备份print(adata.isbacked)# False# 设置备份地址adata.filename ='./test.h5ad'# 查看是否备份成功print(adata.isbacked)# True123456789101112131415当 adata.isbacked状态为 True 后,证明对象已经被写入磁盘;
相反的,我们可以利用 scanpy 很方便地读取文件,获得 AnnData 对象:
importscanpy asscMyadata=sc.read('./test.h5ad')print(Myadata)"""AnnData object with n_obs × n_vars = 1000 × 234 obs: 'time'"""123456783. Scanpy中一些常用api的用法介绍
首先导入Scanpy:
importscanpy assc13.1 sc.pp.filter_cells
sc.pp.filter_cells(data,min_genes=None,max_genes=None)1常常用于预处理中,做一些细胞筛选的工作,该函数保留至少有 min_genes个基因的细胞,或者保留至多有 max_genes个基因的细胞;
另外注意,参数 min_genes和参数 max_genes不能同时传递;
实例:
# 导入数据adata=sc.datasets.krumsiek11()# 5类细胞, 640个细胞样本, 共测量11种基因print(adata)"""AnnData object with n_obs × n_vars = 640 × 11 obs: 'cell_type' uns: 'iroot', 'highlights'"""print(adata.n_obs)# 640个细胞# 11个基因(即特征)print(adata.var_names)"""Index(['Gata2', 'Gata1', 'Fog1', 'EKLF', 'Fli1', 'SCL', 'Cebpa', 'Pu.1', 'cJun', 'EgrNab', 'Gfi1'], dtype='object')"""### 注意观察细胞数量变化 ###sc.pp.filter_cells(adata,min_genes=0)# 相当于没有筛选print(adata.n_obs)# 640print(adata.obs)""" cell_type n_genes0 progenitor 9.. ... ...159 Neu 8细胞一共就5类, 每一类有不同数量个细胞, cell_type左边的index不是dataframe的真正int型index, 是字符串index, 仅表示每类细胞的index, 比如Neu范围是0到159 """print(set(adata.obs['cell_type'].values))# 5类细胞{'Neu', 'progenitor', 'Ery', 'Mo', 'Mk'}print(adata.obs['n_genes'].min())# 4, 每个细胞至少测量了4个基因sc.pp.filter_cells(adata,min_genes=6)# 选择测量了6个基因以上的细胞print(adata.n_obs)# 630print(adata.obs['n_genes'].min())# 61234567891011121314151617181920212223242526272829303132333435363738393.2 sc.pp.filter_genes
sc.pp.filter_genes(data,min_cells=None,max_cells=None)1该函数用于保留在至少 min_cells个细胞中出现的基因,或者保留在至多 max_cells个细胞中出现的基因;
参数 min_cells和参数 max_cells不能同时传递;
对比 sc.pp.filter_cells可以发现,sc.pp.filter_genes用于选择基因(筛选列),sc.pp.filter_cells用于选择细胞(筛选行);
3.3 sc.pp.highly_variable_genes
sc.pp.highly_variable_genes(data,n_top_genes=None,min_disp=0.5,max_disp=inf,min_mean=0.0125,max_mean=3)1234567该函数用于确定高变基因;
常用参数说明:
data:AnnData Matrix,行对应细胞列对应基因n_top_genes:要保留的高变基因的数量
高变异基因就是highly variable features(HVGs),就是在细胞与细胞间进行比较,选择表达量差别最大的基因,Seurat使用FindVariableFeatures函数鉴定高变基因,这些基因在不同细胞之间的表达量差异很大(在一些细胞中高表达,在另一些细胞中低表达)。
3.4 sc.pp.normalize_total
sc.pp.normalize_total(adata,target_sum=None,inplace=True)1函数可以对每个细胞进行标准化,以便每个细胞在标准化后沿着基因方向求和具有相同的总数target_sum;
实例:
adata.Xarray([[3.,3.,3.,6.,6.],[1.,1.,1.,2.,2.],[1.,22.,1.,2.,2.]],dtype=float32)# 设置 target_sum=1 标准化后X_normarray([[0.14,0.14,0.14,0.29,0.29],[0.14,0.14,0.14,0.29,0.29],[0.04,0.79,0.04,0.07,0.07]],dtype=float32)123456789ta.X
array([[ 3., 3., 3., 6., 6.],
[ 1., 1., 1., 2., 2.],
[ 1., 22., 1., 2., 2.]], dtype=float32)
设置 target_sum=1 标准化后
X_norm
array([[0.14, 0.14, 0.14, 0.29, 0.29],
[0.14, 0.14, 0.14, 0.29, 0.29],
[0.04, 0.79, 0.04, 0.07, 0.07]], dtype=float32)
123456789
1. Scanpy简介与安装
Scanpy 是一个可扩展的工具包,用于分析与 AnnData(一种数据结构)联合构建的单细胞分析数据。默认情况下,会返回2,000个高变基因用于下游的分析。
利用FindVariableFeatures函数,会计算一个mean-variance结果,也就是给出表达量均值和方差的关系并且得到top variable features,这一步的目的是鉴定出细胞与细胞之间表达量相差很大的基因,用于后续鉴定细胞类型。
